Diagnóstico genético de la encefalopatía epiléptica de inicio temprano tipo brote supresión [Reseña]
Autores: I Trigo-Pérez y FJ Esteban. Universidad de Jaén
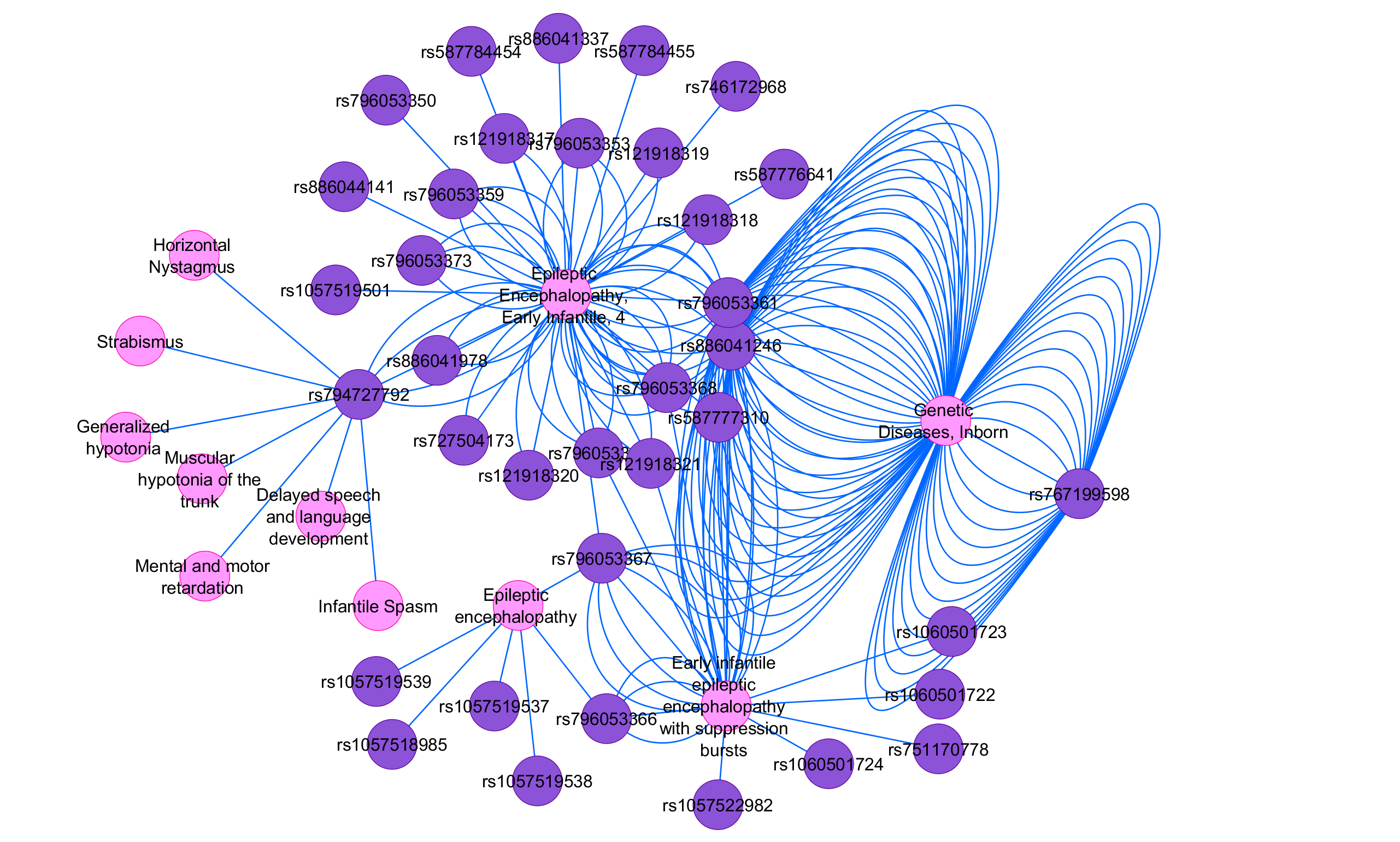
La encefalopatía epiléptica de inicio temprano tipo brote supresión (EOEE-BS) engloba a un grupo de síndromes epilépticos que se caracterizan porque no suelen responder al tratamiento, aparecen durante los tres primeros meses de vida y presentan un patrón encefalográfico de brote supresión. Además, los pacientes con EOEE-BS manifiestan un retraso psicomotor severo.
Dentro de este tipo de encefalopatía epiléptica se encuentran el síndrome de Ohtahara y la encefalopatía mioclónica temprana. La primera suele asociarse a malformaciones de la corteza cerebral, y la segunda a alteraciones congénitas del metabolismo. No obstante, aproximadamente un 60% de los pacientes diagnosticados con EOEE-BS no suelen mostrar anomalías en la estructura de su cerebro y, por otro lado, en el 25% de los casos las malformaciones están asociadas a variantes genéticas.
Por lo anteriormente expuesto, se considera importante llevar a cabo un diagnóstico genético preciso de la EOEE-BS, tanto si se presentan alteraciones estructurales como si no. Sin duda, los recientes avances en secuenciación genética ya lo hacen posible.
En este sentido, recientemente se ha publicado el trabajo “Genetic diagnosis and clinical characteristics by etiological classification in early-onset epileptic encephalopathy with burst suppression pattern”, en el que Sangbo Lee y colaboradores han identificado variantes genéticas asociadas a la EOEE-BS.
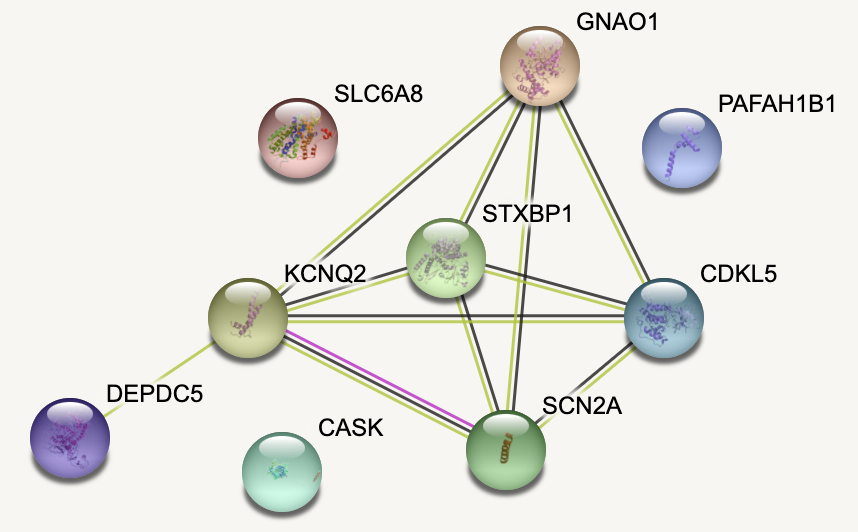
En el estudio se incluyeron un total de 48 pacientes con patología EOEE-BS, pero solo se detectó una alteración genética en 31 de ellos (64.6%). El gen más diagnosticado, en trece casos, fue STXBP1. A continuación, KCNQ2 y SCN2A se detectaron cada uno en cinco pacientes. DEPDC5 en tres. Y CASK, CDKL5, GNAO1 y SLC6A8 en un solo caso en diferentes personas. También se detectó un paciente con deleción en el gen LIS1.
Si nos centramos en los trece participantes en los que se hallaron mutaciones en el gen STXBP1, a once de ellos se les había diagnosticado previamente padecer el síndrome de Ohtahara y, a los otros dos, una encefalopatía mioclónica temprana. A todos se les identificaron crisis tónicas o espasmos epilépticos, así como discapacidad intelectual. Nueve de ellos mostraron hipotonía, y dos pacientes manifestaron características autistas. En cuanto a las anomalías estructurales, se detectaron tres casos de atrofia cerebral y dos de microcefalia. En los trece pacientes, los estudios metabólicos fueron normales.
Diez casos diagnosticados como STXBP1 fueron tratados con una dieta cetogénica, de los cuales siete mostraron una disminución en la frecuencia de crisis y en tres de ellos las crisis desaparecieron. A un paciente se le practicó una cirugía cerebral para seccionar su cuerpo calloso, lo cual indujo un estado casi libre de crisis. En cuanto al tratamiento farmacológico, en seis casos se les administró terapia esteroidea, cinco recibieron levetiracetam y tres tomaron ácido valproico, todo lo cual, en general, dio lugar a alguna mejora. Sin embargo, ningún paciente manifestó un avance significativo en sus funciones cognitivas y motoras.
Los autores resaltan, como conclusión, la importancia del diagnóstico genético de la EOEE-BS, independientemente de las posibles malformaciones cerebrales, pues con ello se facilita la elección del tratamiento, el cual puede ser diferente según el gen alterado causante de la enfermedad.