Sináptica
Sináptica. Esta es la semana del STXBP1 y todo está ocurriendo muy rápido. Nos estamos preparando para el primer Acto Benéfico del STXBP1 y nuestra publicación en la revista Neurology examinando las características fenotípicas de 147 pacientes ha estado online desde hace muy poco. STXBP1 es uno de los cinco genes más comunes para las encefalopatías epilépticas y trastornos relacionados del neurodesarrollo. Sin embargo, en contraste con SCN1A, SCN2A, CDKL5, o SCN8A, ha recibido relativamente poca atención por parte de la comunidad de la epilepsia en estos últimos años. Vamos a revisar un gen habitual de la epilepsia que posee más secretos de lo que la mayoría de gente se imagina.
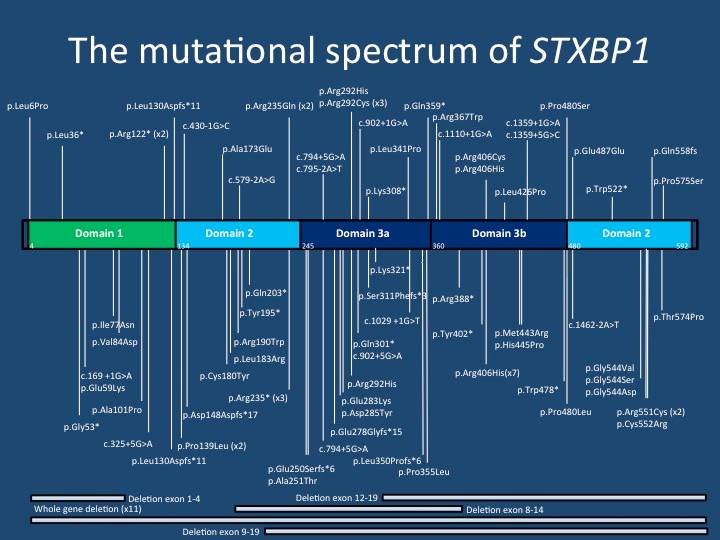
| El espectro mutacional de STXBP1. Esta imagen muestra la mayoría de las mutaciones reportadas en la publicación de Stamberger y sus colaboradores. Las mutaciones se distribuyen en el gen STXBP1, pero hay unos pocos epicentros mutacionales. La mutación p.Arg406His es la mutación más común de novo en el gen STXBP1. |
Adhiérete a STXBP1. Antes de discutir acerca de algunos de los hallazgos en nuestra reciente publicación, déjame enviarte una invitación de última hora para el Acto Benéfico STXBP1 el 20 de febrero en el Club de Campo de Doylestown. Durante el AES 2015 en Filadelfia, tuvimos nuestra primera noche de padres STXBP1 en el CHOP (Children Hospital of Philadelphia) y actualmente estamos en el proceso de construcción de un registro internacional de STXBP1. Por lo tanto, es oportuno que nuestra primera gran publicación STXBP1 apareciera esta semana, que nos proporciona una gran foto panorámica de la situación actual de STXBP1. Para esta publicación, encabezada por Hannah Stamberger como primer autor y Sarah Weckhuysen como autor senior, se extrajeron los datos de los pacientes publicados en el documento y añadimos características fenotípicas de 45 pacientes no reportados previamente.
El fenotipo STXBP1. STXBP1 fue considerado históricamente el gen para el Síndrome de Ohtahara ya que borrados en este gen fueron descubiertos por primera vez en este fenotipo. El gen STXBP1 codifica para una subunidad esencial de la maquinaria de fusión sináptica que permite la transmisión sináptica. Este gen, que también se conoce con el sinónimo Munc-18, es necesario para la transmisión sináptica – ratones en los que se elimina el gen Munc-18 no tienen ninguna transmisión sináptica a pesar de que la estructura del Sistema Nervioso Central se desarrolla normalmente. El mecanismo genético en la encefalopatía STXBP1 es la haploinsuficiencia, es decir, sólo una copia funcional del gen STXBP1 permanece presente en pacientes con encefalopatía STXBP1, ya que la otra copia no produce una proteína funcional debido a una eliminación o mutación. El por qué este estado de haploinsuficiencia aparece en numerosas enfermedades de desarrollo neurológico con epilepsia sigue siendo poco claro. Se supone que las interneuronas se ven más afectadas por esta haploinsuficiencia que las neuronas excitadoras, que a su vez conduce a la hiperexcitabilidad. El cuadro clínico de la encefalopatía STXBP1 siempre ha sido confuso para la comunidad de la epilepsia. Mientras que a algunos pacientes les han sido bien definidos síndromes epilépticos, tales como el Síndrome de Ohtahara o el Síndrome de West, la mayoría de los pacientes tienen un Inicio Temprano de la Encefalopatía Epiléptica (ITEE) que desafía los criterios diagnósticos clásicos, a veces con características de diversos síndromes epilépticos. Estos son los cinco principales mensajes de la publicación de Stamberger que hace hincapié en la complejidad del fenotipo STXBP1.
1 – Los dos dimensiones. Me he centrado en las dos dimensiones de STXBP1 en el título de esta entrada del blog. Básicamente, este concepto se refiere al hecho de que a pesar de que la mayoría de los pacientes de nuestra serie tienen discapacidad intelectual grave, hay muy poca correlación entre la aparición de convulsiones, severidad de las crisis, y el grado de discapacidad intelectual. Esto es algo sorprendente desde una perspectiva clínica. En algunos pacientes con encefalopatía STXBP1, la epilepsia comienza ya en el período neonatal. Sin embargo, el resultado del desarrollo neurológico en estos pacientes no es significativamente diferente de los pacientes que sufren convulsiones que comienzan en una edad más avanzada. En nuestro análisis, no encontramos ninguna correlación entre la «dimensión de la epilepsia» y la dimensión «del desarrollo neurológico».
2 – Dinámica del fenotipo de convulsión. Espasmos epilépticos, convulsiones focales y convulsiones tónicas son los principales tipos de crisis y la mayoría de los pacientes con encefalopatía STXBP1 tienen una actividad epileptiforme multifocal e hipsarritmia. Por lo tanto, la combinación de espasmos epilépticos e hipsarritmia parecen ser la esencia del fenotipo de la encefalopatía STXBP1, a pesar de que existe considerable controversia en torno a este tema. En particular, algunos de las convulsiones focales y EEG multifocales encontrados son a menudo una fuente de confusión en la práctica clínica y pueden sugerir una causa subyacente focal de la epilepsia en la evaluación inicial. Las convulsiones en encefalopatía STXBP1 son altamente dinámicas y comienzan temprano con una media de inicio de 6 semanas, presentándose espasmos en más del 60% de los casos. Sin embargo, un pequeño subgrupo de pacientes tiene otros síndromes epilépticos distintos tales como el Síndrome de Dravet en el 2% de los pacientes. En contraste con muchas otras epilepsias genéticas, más del 40% de los pacientes con encefalopatía STXBP1 dejan de tener convulsiones. Esto está en marcado contraste con el Síndrome de Dravet con mutación en SCN1A, encefalopatía CDKL5, o encefalopatía DNM1.
3 – Otras funciones neurológicas. Hay varias características poco comunes reportadas en algunos pacientes con encefalopatía STXBP1 incluyendo Parkinsonismo juvenil, lo que nos llevó a mirar otras características neurológicas, siempre que esta información estuviera disponible. Se encontró que hipotonía predominantemente axial, ataxia, temblor, espasticidad y distonía eran características en muchos pacientes. Es difícil proporcionar con precisión con qué frecuencia aparece cada característica. También se abordó la cuestión de si la regresión o retroceso es una característica común en la encefalopatía STXBP1, una pregunta clínica habitual que surge cuando se habla sobre el pronóstico con las familias. Sólo encontramos un pequeño subgrupo de pacientes en los que se ha detectado una regresión. Por lo que podemos decir en el 2016, STXBP1 no es una enfermedad neurodegenerativa, pero una encefalopatía estática sí.
4 – Genotipo/fenotipo y mutaciones recurrentes. En total, hemos sido capaces de resumir 123 STXBP1 mutaciones de novo diferentes, con la mayoría de mutaciones únicamente observadas en un solo individuo. Esto proporciona un paisaje mutacional heterogéneo que típicamente se observa en condiciones que resultan de la haploinsuficiencia. En general, no hubo asociación entre el tipo de mutación y grado de discapacidad intelectual o tipo de convulsión. Esto indica que no hay una obvia correlación genotipo/fenotipo de la encefalopatía STXBP1. Además, hay varios epicentros mutacionales en STXBP1. Hemos encontrado 13 mutaciones recurrentes en STXBP1, incluyendo p.R406H en siete pacientes y p.R551C en cuatro pacientes. Los siete pacientes con p.R406H tuvieron convulsiones a partir de 2,5 meses y tenían de severa a profunda discapacidad intelectual. Sin embargo, el rango de fenotipos convulsivos y desarrollo neurológico resultante es más variable para las otras mutaciones recurrentes, incluyendo p.R292C, p.R292H, y p.R551C.
5 – Frecuencia de población. ¿Qué frecuencia tiene la encefalopatía STXBP1? Dada la atención centralizada para los pacientes con epilepsia en Dinamarca, donde se ven prácticamente todos los niños con epilepsias graves en un solo centro, somos capaces de estimar las frecuencias de población de epilepsias genéticas raras. Por ejemplo, la frecuencia de la población para el Síndrome de Dravet se estima que es 1: 22.000. En contraste con el fenotipo relativamente homogéneo del Síndrome de Dravet, la estimación de la encefalopatía STXBP1 es más difícil. Utilizando la misma metodología, llegamos a una frecuencia de población de 1: 91.000. Esto, sin embargo, no toma en cuenta los pacientes con crisis leves que no han sido incluidos en la prueba y no da cuenta de los pacientes con STXBP1 que no presentan convulsiones.
Esto es lo que necesitas saber. La reciente publicación de Stamberger y sus colaboradores es un importante paso adelante en nuestra comprensión de la encefalopatía STXBP1. Ahora tenemos una buena visión general de las características clínicas principales de pacientes con epilepsia y entendemos que, a pesar de que la epilepsia puede comenzar temprano y de manera muy dramática, una proporción significativa de pacientes dejará atrás las convulsiones. Además, hay poca correlación entre la ausencia de crisis y el resultado del desarrollo. La mayoría de los pacientes con encefalopatía STXBP1 tienen discapacidad intelectual grave en nuestra serie. Sin embargo, la regresión y pérdida de habilidades a través del tiempo es una característica poco común. Hay poca correlación genotipo/fenotipo y los pacientes con idénticas mutaciones de novo pueden tener diferentes fenotipos. En general, todo esto hace hincapié en que STXBP1 es algo más que una encefalopatía epiléptica. Es una condición del desarrollo neurológico complejo que requiere de una atención multidisciplinaria.

| Ingo Helbig es miembro de Neurología Infantil e investigador de genética de la epilepsia en el Hospital Infantil de Filadelfia (CHOP), EE.UU. y el Departamento de Neurología Pediátrica, Kiel, Alemania. |

1 Comment
Esta muy bueno articulo acaban de enviarlo ya que hoy diagnosticaron a mi hijo con la.mutación del gen STXBP1
Me gustaría si ustedes pudieran enviarme más información delante mano gracias