Un consorcio europeo coordinado por el investigador Ruud Toonen del CNCR recibe 1 millón de euros para la investigación de la epilepsia.
Buenas noticias! Un consorcio europeo coordinado por el investigador Ruud Toonen del CNCR (Center for Neurogenomics and Cognitive Research) de Amsterdam, recibe 1 millón de euros para la investigación de la epilepsia, en especial la causada por mutaciones en genes responsables de la transmisión sináptica, como el STXBP1.
La epilepsia es una grave e incapacitante enfermedad que afecta aproximadamente al 1% de la población mundial. A pesar de años de intensa investigación, alrededor del 30% de todas las epilepsias no pueden ser tratadas con medicamentos disponibles. Para desarrollar nuevas y mejores opciones de tratamiento, se requiere con urgencia un conocimiento detallado de los mecanismos que conducen a la epilepsia.
Toonen dirigirá el proyecto de investigación transnacional ERA-NET NEURON «SNAREopathy» que se centrará en un grupo de epilepsias severas difíciles de tratar causadas por mutaciones en los genes que median la comunicación entre las células nerviosas (transmisión sináptica). El consorcio reúne a expertos en genética de epilepsia y neurobiólogos que trabajan en la transmisión sináptica de los Países Bajos, Alemania, Italia y Noruega para identificar los mecanismos que conducen a la epilepsia utilizando varios modelos sofisticados de ratón y pez cebra, así como modelos celulares humanos derivados de biopsias de piel que se transformarán en neuronas humanas (llamadas neuronas derivadas de iPSC). Basándose en los mecanismos epilépticos identificados, el consorcio utilizará estos modelos en la detección de sustancias para buscar nuevos medicamentos que traten mejor y más específicamente a los pacientes con epilepsia gravemente afectados.
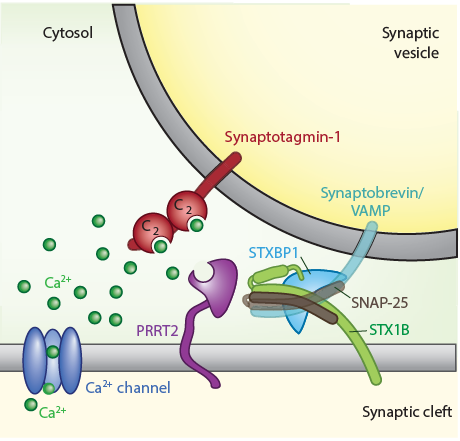
La interacción de SNAREs (Soluble N-ethylmaleimide–sensitive factor-attachment protein receptors) y STXBP1 / MUNC18-1, junto con las proteínas de unión a calcio de la familia synaptotagmin garantiza la velocidad y la precisión de la liberación de neurotransmisores. Es importante destacar que las mutaciones en dos componentes centrales del complejo SNARE y un cofactor, codificados por los genes STX1B, STXBP1 y PRRT2 (véase la figura), conducen a un amplio espectro de fenotipos de convulsiones y otros síntomas neuropsiquiátricos, muchos de los cuales responden mal a medicamentos antiepilépticos disponibles. Curiosamente, los mismos genes están asociados con trastornos del autismo y la esquizofrenia con una mayor comorbilidad convulsiva. Han identificado mutaciones en dos de los tres genes, STX1B y PRRT2, en formas familiares o esporádicas de epilepsia. Similar pero clínicamente distinguibles de las mutaciones STX1B, las mutaciones de novo en STXBP1 causan una epilepsia severa de inicio temprano con un patrón característico de supresión-ráfaga en el EEG. Ahora combinarán su conocimiento de estas proteínas presinápticas para llegar a una imagen integral de la disfunción presináptica en los trastornos epilépticos y las comorbilidades relacionadas usando modelos animales, neuronas derivadas de pacientes e innovadoras plataformas de cribado in vitro e in vivo para desarrollar nuevos enfoques terapéuticos a medida.
Publicación original haciendo click aquí

